The Anti-NMDA Receptor Encephalitis Foundation, Inc.was established in Canada as a not-for profit-foundation on 26 October 201…
|
J Neurol Neurosurg Psychiatry. 2019 Jun;90(6):652-658. doi: 10.1136/jnnp-2018-319714. Epub 2019 Jan 13.
|
Introduction N-methyl-D-aspartate receptor antibody-mediated encephalitis (NMDAR-AbE) is an increasingly recognised and treatable encephalitis, with a predilection for children and young adults.1 2 As earlier immunotherapy improves outcomes, timely and accurate recognition of NMDAR-AbE is a major clinical aim.2 The characteristic polysymptomatic presentation of NMDAR-AbE includes early neuropsychiatric deficits with seizures, autonomic disturbance, reduced consciousness and a movement disorder (MD).1 2 This MD, seen in around 90% of cases, can be the presenting feature, particularly in children,1 and is typically hyperkinetic with limb plus orofacial involvement.1–5 To date, elegant detailed descriptions exist in a few patients.4 However, small series have used highly variable phenomenological descriptions.1–5 Therefore, the phenomenology of the associated MD lacks consensus. Its clearer description will facilitate confident recognition and enable earlier immunotherapy administration in NMDAR-AbE. Expert-rater descriptions remain the gold standard to define phenotypes in movement disorders. In this study, ratings from seven experts across 76 videos were used to better define the MD in NMDAR-AbE. Methods Subjects Autoimmune neurology researchers contributed 44 videos from 20 patients who met diagnostic criteria for NMDAR-AbE.e1 A PubMed search for ‘anti-NMDAR encephalitis’ and ‘NMDAR-antibody encephalitis’, plus associated references, revealed 32 videos from 14 subjects, in eight papers from 14 subjects.3 , e2–8 Clinical features, temporal progression, outcomes and investigation findings were collated from case note reviews by the researchers and from data in published papers. Twenty videos from 18 age-matched and sex-matched (age range 2–41 years, median 12, 50% female) disease controls were selected from the literature (online supplementary data).3 e9–19 Supplementary data Movement disorder classification Seven experts (KB, VF, AEL, TL, NN, KS and MT) established a consensus glossary of terms (online supplementary data, modified from Mohammed et al 3). Subsequently, each expert blindly and independently rated 76 videos from 34 patients with NMDAR-AbE (median two videos per patient, range 1–7) plus 20 disease controls, using glossary-derived terms within a standardised data collection sheet (online supplementary data). Results Cohort characteristics From the 34 patients, median age was 7 years (range 0.2–32 years) and 59% were female. Consistent with this young cohort, 6% (2/34) had ovarian teratomas. Peak modified Rankin Scale showed a severe illness (median 5; range 2–5), and, other than the prerequisite MD, the clinical features and investigation findings were in accordance with other series of patients with NMDAR-AbE (figure 1A–B).1 2 Overall, the MD was an early feature of the disease (figure 1B) and, in addition to the known prolonged psychiatric symptoms, persisted for a median of 112.5 days (range 30–1482, figure 1C). Figure 1 The clinical features and movement disorder evaluations in 34 patients with N-methyl-D-aspartate-antibody encephalitis (NMDAR-AbE). (A) Clinical and investigation findings across the 34 patients whose videos were rated. By definition, all patients had a movement disorder (MD) and other clinical and paraclinical features included psychiatric (n=33/34), cognitive (n=32/34), seizures (n=30/34), autonomic (n=17/34), abnormal electroencephalogram (n=28/30), abnormal cerebrospinal fluid (CSF) (n=19/33) and abnormal MRI (n=9/34). Abnormal CSF findings included any of pleocytosis, oligoclonal bands or raised protein. Two ovarian teratomas were noted in postpubescent women (25 and 32 years). Overall, three cases were adults. (B) Symptom onset and offset in NMDAR-AbE: timings of onset (black) and offset (grey) of the main five symptom categories after first symptom (day 1; median, minimum and maximum values displayed on box and whisker plot), (C) with a particular focus on the timing of the MD in individual patients. Full datasets were available from coauthors and denominators <34 represent variably reported details from the literature-derived videos. Expert classification of phenomenology for 76 videos from patients with NMDAR-AbE (D–F). (D) Dystonia, chorea and stereotypies were the most commonly used terms. For the ‘other’ category, raters used terms including: mutism, stupor, myorhythmia, myokymia, tics, opisthotonus, cerebellar syndrome/ataxia, orofacial dyskinesia, waxy flexibility, oculogyric crises, athetosis, agitation, seizure, startle and vocal perseveration. (E) The interactions between phenomenologies in NMDAR-AbE with co-occurrence of stereotypies, chorea and dystonia shown in a Circos plot,e20 based on a co-occurrence matrix within single video ratings. (F) Stereotypies, chorea and dystonia were equally represented in the face, arm and leg, respectively. Evaluation of videos Videos were recorded at a median of 28 days from symptom onset (range 1–690 days). Across all videos, three principle dominant MDs were noted (figure 1D): dystonia (20%), chorea (20%) and stereotypies (19%). Other hyperkinetic phenomenologies included clonic perseveration (6%), ballism (6%), myoclonus (5%), tonic perseveration (3%) and tremor (1%). Hypokinetic terms included catatonia (6%) and bradykinesia (5%). Three representative videos are presented in the online supplementary video 1-3. Overall, fewer phenomenological terms were used to describe the MDs from disease controls (mean 1.9 per video, range 1–4; online supplementary data) versus patients with NMDAR-AbE (mean 3.4 per video, range 1–7; p<0.0001, Mann-Whitney U-test). This complexity was consistent with the strong inter-rater reliability across the 20 disease control videos in comparison to the 76 NMDAR-AbE videos, for the dominant phenomenology as dystonia (κ=0.65 vs 0.39, controls vs NMDAR-AbE), chorea (κ=0.63 vs 0.39) or stereotypies (κ=0.91 vs 0.22). Analysis of the frequency of MDs within a single video in the patients with NMDAR-AbE revealed marked coexistence of chorea, dystonia and stereotypies (figure 1E and online supplementary figure 1). Indeed, 97% (33/34) of patients and 79% (60/76) of videos contained two of dystonia, chorea and stereotypies, with 76% (26/34) of patients and 46% (35/76) of videos containing all three phenomenologies. Overall, very similar patterns were observed with inclusion of associated disorders (online supplementary figure 2)3 and across ages and gender (data not shown). Temporal variability and regional distribution of the movement disorder To explore these patterns over time, several patients were rated longitudinally. Dystonia, stereotypies and chorea remained prominent but there was marked variability in the MD within an individual patient over the course of a single day and over a number of weeks (median=21 days, range 0–360 days; online supplementary figure 3). Finally, the anatomical distribution of these phenomenologies revealed similar ratings in the face, arm and leg (figure 1F). Discussion NMDAR-AbE is a severe yet treatable neurological disorder in which prompt administration of immunotherapies improves outcomes.2 The MD is a common and well-emphasised feature of disease descriptions, yet until now lacked an expert-based gold-standard description.1–5 From this study, the MD was an early and persistent feature and, from a large collection of videos, it was characterised by diverse, widely distributed hyperkinetic phenomenologies, with prominent dystonia, chorea and stereotypies, with almost complete absence of tremor and tics and a relative paucity of myoclonus. Furthermore, it was complex to describe using conventional MD terminology, yet the above features may help differentiate this mixed movement disorder from important differential diagnoses in routine clinical practice including primary psychiatric disorders, childhood metabolic disturbances and drug intoxication. Indeed, the coexistence of widely distributed chorea, dystonia and stereotypies is rare in other neurological conditions (expert panel, unpublished observations) and may reflect the targeted autoantibody-mediated downregulation of NMDARs. In this study, expert-rater feedback stated ‘our definition and description of hyperkinetic movements do not cover some of these patients ’and ‘reviewing all of these videotapes was a challenge particularly to preconceived and established phenomenological categorisations. Not infrequently I was dissatisfied with the final category that I chose’. This difficulty in classifying patients with conventional terms is reflected in the inter-rater kappa statistics and the greater variety of terms used to describe patients with NMDAR-AbE. These kappa values could be interpreted as reflecting variability between expert-raters, but they showed good agreement across other varied control disorders and many terms are not interchangeable. However, potential study limitations include the lack of accompanying clinical histories and standardised video assessments, the overall young age and marked disease severity, the surprisingly prolonged duration of the MD and an intrinsic bias towards recording the most unusual movements. Future extension studies could also incorporate a consecutive series of patients with a more exhaustive list of control mixed movement disorders. In summary, this clinical constellation of phenomenologies, with the unusual triad of dystonia, stereotypies and chorea, should, in the correct clinical context, provide clinicians with greater confidence in diagnosing this common cause of encephalitis, allowing earlier immunotherapy administration and improved patient outcomes. Supplementary video Supplementary video Supplementary video References ↵ Florance NR , Davis RL , Lam C , et al . Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol 2009;66:11–18.doi:10.1002/ana.21756 ↵ Irani SR , Bera K , Waters P , et al . N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010;133(Pt 6):1655–67.doi:10.1093/brain/awq113 ↵ Mohammad SS , Fung VS , Grattan-Smith P , et al . Movement disorders in children with anti-NMDAR encephalitis and other autoimmune encephalopathies. Mov Disord 2014;29:1539–42.doi:10.1002/mds.25999 OpenUrlCrossRefPubMed ↵ Kleinig TJ , Thompson PD , Matar W , et al . The distinctive movement disorder of ovarian teratoma-associated encephalitis. Mov Disord 2008;23:1256–61.doi:10.1002/mds.22073 ↵ Baizabal-Carvallo JF , Stocco A , Muscal E , et al . The spectrum of movement disorders in children with anti-NMDA receptor encephalitis. Mov Disord 2013;28:543–7.doi:10.1002/mds.25354 OpenUrlCrossRefPubMed
|
Anti-N-methyl-D-aspartate receptor encephalitis is a recently identified autoimmune disorder with a prominent neuropsychiatric presentation. We presen..
|
For the first time specific antibodies have been found to be associated with the onset of schizophrenia in a study led by Professor Belinda Lennox.
|
|
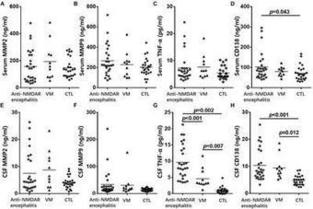
Background: Syndecan-1 is an important component of endothelial cell glycocalyx, and has been reported to be a negative regulator of various inflammatory processes. The clinical implication of circulating and CSF syndecan-1 (sCD138) in patients with anti-N-methyl-D-aspartate receptor (NMDAR)…
|
We are Anti NMDA Receptor Encephalitis! | We are Anti NMDA Receptor Encephalitis!From…
|
I screamed her name. I rubbed her chest. I rushed her to the ER. It took a long time before we got the autoimmune encephalitis diagnosis.
|

KEY POINTS Autoimmune limbic encephalitis is an inflammatory disease involving the medial temporal lobes; it classically presents with the subacute onset of short-term memory deficits, seizures or psychiatric symptoms. Brain magnetic resonance imaging can show medial temporal lobe abnormalities typical of autoimmune limbic encephalitis in suspected cases, but clinicians should be aware of other diseases that may have a similar imaging appearance. Analysis of both electroencephalogram (EEG) and cerebrospinal fluid can provide supportive evidence of neuro-inflammation in patients with suspected autoimmune limbic encephalitis, but a normal EEG or cerebrospinal fluid profile does not exclude the diagnosis. Testing for antibodies to onconeural, cell-surface and synaptic proteins represents a major advancement in the diagnosis of autoimmune limbic encephalitis, although false-positives are possible. Autoimmune limbic encephalitis (ALE) is an inflammatory disease involving the medial temporal lobes; it classically presents with rapid neuropsychiatric decline. Patients with ALE have, and may present with, a diverse array of neuropsychiatric symptoms, which means that they may initially be assessed by any one of a range of medical practitioners. The condition was first described as a paraneoplastic phenomenon, but subsequently, with the discovery of disease-causing antibodies, was shown to be nonparaneoplastic in many cases.1,2 Although ALE is uncommon (an epidemiologic study of encephalitis found the prevalence of ALE without antibody positivity to be only 2 cases per 100 000 people), the incidence of autoimmune encephalitides has risen over the last decade, driven largely by improved antibody detection.3 Autoimmune limbic encephalitis is commonly misdiagnosed, yet early diagnosis and treatment can improve outcomes. We review the approach to diagnosis of ALE, drawing on findings of large cohort and case-control studies, which represent the highest level of evidence in this field (Box 1). We discuss diagnostic criteria for ALE presented in a 2016 position paper by Graus and colleagues (hereafter referred to as the Graus criteria; Box 2),4 which aim to prevent overdiagnosis of ALE and are highly specific.5 These criteria serve as an excellent resource for both specialists and generalists. The treatment of ALE is best coordinated by a specialist in autoimmune neurologic diseases and is beyond the scope of this review. Box 1: Search strategy for this review We screened titles of all publications via PubMed pertaining to the diagnosis of autoimmune limbic encephalitis and/or exclusion of its mimics, dating back to 1964, and reviewed those that were relevant to the subject. We also reviewed the references of all relevant publications for potential inclusion. We emphasized larger cohort and case-control studies over case reports, as well as publications within the last 10 years, given the recent advancements in antibody testing. Box 2: Diagnostic criteria for definite autoimmune limbic encephalitis4 Diagnosis can be made when all 4* of the following criteria have been met: Subacute onset (rapid progression of less than 3 mo) of working memory deficits, seizures, or psychiatric symptoms suggesting involvement of the limbic system. Bilateral brain abnormalities on T2-weighted fluid-attenuated inversion recovery MRI highly restricted to the medial temporal lobes† At least one of the following: CSF pleocytosis (white blood cell count of more than 5 cells per mm3) EEG with epileptic or slow-wave activity involving the temporal lobes Reasonable exclusion of alternative causes Reprinted from Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 2016;15:391–404, with permission from Elsevier. Note: CSF = cerebrospinal fluid, EEG = electroencephalogram, MRI = magnetic resonance imaging. ↵* If one of the first 3 criteria is not met, a diagnosis of definite limbic encephalitis can be made only with the detection of antibodies against cell-surface, synaptic, or onconeural proteins. ↵† 18 Fluorodeoxyglucose (18 F-FDG) PET can be used to fulfil this criterion. Results from studies from the past 5 years suggest that 18 F-FDG-PET imaging might be more sensitive than MRI to show an increase in FDG uptake in normal-appearing medial temporal lobes. What are the clinical features of autoimmune limbic encephalitis? Typical symptoms of ALE reflect dysfunction of the limbic structures of the brain and include short-term memory deficits, behavioural changes, anxiety, depression, psychosis and seizures. 1,6,7 Autoimmune limbic encephalitis most often occurs in middle-aged adults, but it can affect people of all ages, ranging from children to older people.1,6–8 Pace of disease progression In retrospective studies of patients with ALE, the median time from symptom onset to clinical assessment was usually several weeks;6,9 this subacute presentation of the disease is highlighted in the Graus criteria and is a hallmark of the disorder.4 Although ALE should be considered in any patient who presents with rapidly progressive memory difficulties, behavioural changes, psychiatric symptoms or seizures of unclear cause, an individual who presents with sudden-onset neurologic symptoms is more likely to have suffered an acute neurologic or systemic insult such as a stroke or toxic ingestion. However, it is important not to classify a patient’s illness as acute before questioning family, friends or caregivers about subtle memory problems or behavioural changes in the preceding days or weeks. Conversely, an individual may seem to have a precipitous deterioration concerning for ALE, but after further history-taking, it becomes apparent that there has been milder cognitive impairment over many months, or even years. Although certain antibodies have been associated with a more insidious presentation of ALE,10 a protracted disease course should alert the physician to the possibility of an alternative diagnosis such as a neurodegenerative disease. Involuntary movements Clinicians should ask caregivers about any hemi-body jerking leading up to presentation, which may represent faciobrachial dystonic seizures. These involuntary movements consist of brief contractions that affect the ipsilateral face, arm and sometimes leg; last a few seconds; can occur up to hundreds of times a day; and are often refractory to treatment with anti-epileptic drugs.11 They are seen with anti–leucine-rich glioma inactivated 1 (LGI1) antibodies, the most common antibody causing ALE.9 Other symptoms likely resulting from focal seizure activity may occur in anti-LGI1 encephalitis, including thermal sensations, piloerection and paroxysmal dizziness spells, but faciobrachial dystonic seizures are especially helpful diagnostically as they are nearly pathognomonic of this disease.11–14 Importantly, a normal electroencephalogram (EEG) during faciobrachial dystonic seizures should not dissuade practitioners, as coincident epileptiform discharges are seen only in the minority.11 A retrospective study of 26 patients with such seizures found that they preceded cognitive impairment typical of ALE in 20 patients (77%);11 thus, faciobrachial dystonic seizures may be a clue to prodromal autoimmune neurologic disease in a patient who appears to present acutely. Why is prompt, correct diagnosis essential? A retrospective study examining admission diagnoses of 50 patients with autoimmune encephalitides such as ALE found that two-thirds were initially thought to have conditions other than encephalitis, including primary psychiatric disorders, idiopathic epilepsy, cerebral ischemia or neurodegeneration.15 Even among the one-third of patients in whom encephalitis was considered, an infectious rather than autoimmune cause was more commonly assumed.15 The potential symptom overlap between these 2 disease processes was highlighted in a prospective study of 203 patients with encephalitis, which found that many traditionally infectious symptoms such as fever did not readily distinguish between an infectious and immune-mediated cause.16 Identification of ALE is important because it facilitates prompt use of immunotherapy which, in observational studies, has been associated with reduced seizure frequency, recovery of cognition and likely even improved survival.12,17 Recognition of ALE also triggers malignancy screening, especially among patients with antibodies that strongly predict the presence of a tumour (discussed later; Table 1). Detection of any occult neoplasm is critical as the malignancy may ultimately determine clinical outcome.1 Table 1: Antibodies that may be found in autoimmune limbic encephalitis, and their tumour associations What tests aid in the diagnosis of ALE? Magnetic resonance imaging Magnetic resonance imaging (MRI) of the brain can show medial temporal lobe changes typical of the disease (Figure 1) and is recommended by expert consensus in suspected cases.4 18-Fluorodeoxyglucose positron emission tomography (18-FDG PET) of the brain may be even more sensitive for temporal lobe abnormalities;29 because this modality is not as accessible to many Canadian practitioners, MRI remains a first-line neuroimaging technique. Bilateral imaging abnormalities restricted to the medial temporal lobes are required for a definitive diagnosis of ALE in the absence of antibody positivity. It is important to bear in mind that other diseases — such as other infectious and inflammatory encephalitides, as well as vascular or neoplastic conditions — may involve these structures (Appendix 1, available at www.cmaj.ca/lookup/suppl/doi:10.1503/cmaj.181548/-/DC1). Figure 1: Brain magnetic resonance imaging in autoimmune limbic encephalitis. On coronal fluid-attenuated inversion recovery (FLAIR) image, bilateral T2-hyperintensity of the medial temporal lobes is seen (A, arrows). (B) Bilateral T2-hyperintensity of the medial temporal lobes is also shown on an axial FLAIR image (arrows). Antibodies targeting contactin-associated protein-like 2 were identified in serum by cell-based assay. Differential diagnosis of temporal lobe changes on MRI In a retrospective review of 251 suspected cases of encephalitis with temporal lobe abnormalities on MRI, nearly 25% were due to herpes simplex virus encephalitis; it is therefore important to rule out this potentially devastating infection in patients with suspected ALE.30 Unilateral rather than bilateral temporal lobe changes, insular involvement and absence of basal ganglia involvement are neuroimaging clues that suggest herpes simplex virus encephalitis rather than ALE (Figure 2A, 2B).30,31 Other major infectious considerations are varicella zoster virus, tuberculosis and neurosyphilis; appropriate testing for these entities should be considered early in the disease course.30,32 Figure 2: Magnetic resonance imaging (MRI) mimics of autoimmune limbic encephalitis. (A, B) Herpes simplex virus encephalitis: On axial T2-weighted image, diffuse T2-hyperintensity of the right anteromedial temporal lobe with swelling is seen (A, arrow). There is also T2-hyperintensity of the right insula (B, arrow) that spares the adjacent basal ganglia (red line demarcating separation). (C, D) Temporal lobe glioma: On axial T2-weighted image, diffuse T2-hyperintensity of the right anteromedial temporal lobe is seen (C, arrow). On follow-up MRI 6 months later, there is heterogenous enhancement with local edema on axial T1-weighted post-gadolinium image, concerning for transformation into high-grade glioma (D, arrow). (E) Status epilepticus: On axial T2-weighted image, there is focal T2-hyperintensity of the medial temporal lobes bilaterally (arrows). (F) Acute ischemic stroke: On axial diffusion–weighted image, there is a diffusion-bright lesion of the left medial temporal lobe within the territory of the left posterior cerebral artery (arrow). There is no extension of the lesion into the more anterior or lateral temporal lobe supplied by the left middle cerebral artery (red line demarcating separation), in keeping with acute infarction restricted to a vascular territory. Several noninfectious diseases may involve the temporal lobes and be mistaken for ALE. Gliomas may cause diffuse temporal lobe changes on MRI, while imaging features of high-grade neoplasm such as necrosis, irregular enhancement and vasogenic edema are absent early on. Although classically thought to present unilaterally, in one retrospective series, bilateral medial temporal lobe involvement was seen in 54% cases of patients with suspected ALE who later developed glioblastoma.33 Follow-up with magnetic resonance imaging is therefore recommended in any patient with possible ALE who progresses atypically, as malignant transformation indicative of high-grade neoplasm may occur (Figure 2C, 2D). Seizures can also cause temporal lobe imaging abnormalities34 (Figure 2E), but early control of seizures with anti-epileptic drugs alone, lack of prodromal neuropsychiatric symptoms and the resolution of temporal lobe changes after cessation of seizure activity are all supportive of seizure-related MRI changes rather than ALE. Ischemic stroke involving the medial temporal lobe usually presents acutely but sometimes causes only mild neurocognitive deficits; patients may delay seeking medical attention. Careful history-taking is needed to differentiate a subacute progression of symptoms over days from a static neurologic insult that occurred days earlier. On MRI, signal abnormality restricted to a vascular territory helps distinguish ischemic stroke from ALE35 (Figure 2F). Electroencephalography Patients with ALE may occasionally have a normal EEG, which does not rule out the diagnosis.9,36 Usually, however, EEG shows slow-wave activity or epileptiform discharges from the temporal lobes of patients with ALE — a clue to brain inflammation. In a retrospective analysis of 19 patients with autoimmune encephalitis and seizures, 10 of 16 patients with ictal EEGs (63%) had seizure onset over the temporal lobe region, which closely mirrored the medial temporal lobe abnormalities seen on MRI in three-quarters of those studied.37 Importantly, EEG abnormalities of the temporal lobes without any observed changes on imaging are not sufficient to make a diagnosis of ALE in the absence of a positive antibody, according to the Graus criteria4 outlined in Box 2. In clinical practice, an EEG that shows slow-wave activity or epileptiform discharges from the temporal lobes in a patient with possible ALE should raise suspicion of the condition even if the initial MRI is normal; in such cases, repeat MRI may be considered, to look for the interval development of medial temporal lobe abnormalities.38 Cerebrospinal fluid analysis Analysis of cerebrospinal fluid can provide evidence of neuro-inflammation in patients with possible ALE. In a retrospective study of 50 patients with paraneoplastic ALE, about 50% had a modest leukocyte pleocytosis in cerebrospinal fluid (< 100 cells/μL), while three-quarters of samples tested for oligoclonal bands were positive. 1 In a larger retrospective pooled-data analysis of 205 patients with ALE, however, leukocyte pleocytosis and oligoclonal bands were each noted in only about 25% of tested cerebrospinal fluid samples.39 These findings suggest that although the presence of leukocyte pleocytosis and oligoclonal bands in cerebrospinal fluid supports a diagnosis of ALE in the appropriate clinical context, their diagnostic sensitivity is low. This is reflected in the Graus criteria, which do not require leukocyte pleocytosis or oligoclonal bands in cerebrospinal fluid for a diagnosis of definite ALE even in the absence of antibody positivity. Testing of the cerebrospinal fluid is also helpful to exclude mimics of ALE, in particular herpes simplex virus encephalitis. A retrospective study found that the cerebrospinal fluid profile alone (leukocyte count, erythrocyte count, protein and glucose) could not reliably differentiate between herpes simplex virus encephalitis and autoimmune encephalitis with temporal lobe abnormalities.30 Polymerase chain reaction (PCR) testing for herpes simplex virus in the cerebrospinal fluid has high diagnostic sensitivity and specificity, but clinicians should be aware that it may be negative early in the disease course.40 If a patient has a negative PCR result but herpes simplex virus encephalitis remains a concern clinically or on neuroimaging, continuing antiviral treatment while awaiting a second lumbar puncture for repeat PCR testing is wise.40 Antibody testing Testing for antibodies to onconeural, cell-surface or synaptic proteins is very helpful in suspected ALE, as a positive disease-specific antibody can make the diagnosis in patients who would not otherwise satisfy Graus criteria.4 The recognition that ALE may be associated with malignancy was followed by the discovery of antineuronal antibodies that supported an immune-mediated disease mechanism.1,41 In a retrospective study of paraneoplastic ALE, antibodies to neural antigens expressed by a tumour (referred to as onconeural antibodies) were identified in 30 of 50 (60%) of patients.1 These were most often anti-Hu or Ma2 antibodies, which are classically found with small-cell lung cancer and testicular tumour, respectively.1 Onconeural antibodies bind intracellular antigens and are therefore of unclear pathogenic significance in ALE; they may simply be an epiphenomenon of a primarily cytotoxic T-cell–mediated process that can lead to irreversible neuronal damage and poor clinical outcomes.42,43 Antibodies to the intracellular antigen glutamic acid decarboxylase may also be associated with ALE, but at much higher titres than are typically seen in type 1 diabetes mellitus.44 More recently, antibodies targeting neuronal cell-surface or synaptic proteins have been discovered in patients with ALE; they bind extracellular antigens and are thus more likely to be pathogenic.6,7,45 These antibodies are variably associated with malignancy, and patients often improve with immunotherapy, owing to reversal of antibody-mediated neuronal dysfunction.2 In a retrospective study of 163 patients with ALE, antibodies were found in 93%, the majority of which targeted neuronal cell-surface or synaptic proteins: LGI1 in 44%, γ-aminobutyric acid B receptor (GABABR) in 16%, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) in 7%, and contactin-associated protein-like 2 (CASPR2) in 6%.9 Of note, antibodies to the voltage-gated potassium channel were initially reported in ALE but later found to target the associated proteins LGI1 and CASPR2, rather than the voltage-gated potassium channel itself.46 Antibodies to the N-methyl-d-aspartate receptor (NMDAR) may be identified in ALE, but more often are found in patients with a normal MRI and a characteristic clinical syndrome (anti-NMDAR encephalitis), consisting of abnormal behaviour, speech dysfunction, seizures, dyskinesias or dysautonomia.20,47 Approach to antibody testing Both serum and cerebrospinal fluid testing for the most commonly identified antibodies in ALE (anti-LGI1, GABABR, AMPAR, CASPR2, Hu, Ma2 and GAD) should be considered, to maximize diagnostic yield, as some antibodies (e.g., anti-LGI1) are more sensitive in serum and others (e.g., anti-GABABR) may be identified only in cerebrospinal fluid.14,17 Antibody testing is also worthwhile even in a patient who already meets Graus criteria for definite ALE, as a positive antibody may indicate the likelihood of a specific tumour (Table 1) and inform malignancy screening. The presence of an antibody with a strong tumour association should prompt repeated screening for malignancy if an initial screen is negative, to ensure an occult neoplasm is not missed.48 Even if a patient is ultimately determined to have a diagnosis other than ALE, a positive antibody still requires investigation if it strongly associates with an underlying tumour (e.g., anti-Hu).49 Further detail regarding screening for tumours in paraneoplastic neurologic syndromes such as ALE is beyond the scope of this review, but a comprehensive guideline has been published.48 If a positive antibody is reported in a patient deemed unlikely to have ALE, inquiry should be made into whether the testing laboratory can perform a confirmatory assay to exclude false-positives.10,50 Conclusion Accurate diagnosis of ALE is critical to ensure appropriate management of the disease and to maximize the likelihood of a good patient outcome. Although recently published criteria provide a valuable diagnostic framework for ALE, it is important to understand the rationale behind using conventional diagnostic tools (MRI, EEG and analysis of cerebrospinal fluid), as well as their limitations. While questions remain (Box 3), the ongoing discovery of antibodies to onconeural, cell-surface and synaptic proteins represents a major advancement in the refined diagnosis of ALE, and, as such testing becomes more accessible, a thoughtful diagnostic approach will help to balance patient care with resource management in Canada. Box 3: Unanswered questions Should 18-fluorodeoxyglucose positron emission tomography routinely be performed in patients with suspected autoimmune limbic encephalitis who have a normal magnetic resonance imaging scan? Should more extensive antibody testing be performed in patients who already meet criteria for definite autoimmune limbic encephalitis, but are negative for the most common antibodies associated with this disease? What is the threshold of suspicion for autoimmune limbic encephalitis above which antibody testing should be sent, so as to avoid indiscriminate ordering and minimize the risk of false-positive results? Should methods of antibody testing be standardized across Canadian institutions? Footnotes Competing interests: Jorge Burneo reports receiving grants from the Rick Berg Legacy Fund, the Ontario Brain Institute and Western University, and other support through the Jack Cowin Chair in Epilepsy Research (a Western University Research Chair), outside the submitted work. No other competing interests were declared. This article has been peer reviewed. Contributors: All of the authors contributed to the conception and design of the work, and the acquisition, analysis, and interpretation of data. Adrian Budhram drafted the manuscript. Andrew Leung, Michael Nicolle and Jorge Burneo revised it critically for important intellectual content. All authors gave final approval of the version to be published and agreed to be accountable for all aspects of the work. References ↵Gultekin SH, Rosenfeld MR, Voltz R, et al. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients. Brain 2000;123:1481–94.OpenUrlCrossRefPubMed ↵Dalmau J, Graus F. Antibody-mediated encephalitis. N Engl J Med 2018;378:840–51.OpenUrlCrossRefPubMed ↵Dubey D, Pittock SJ, Kelly CR, et al. Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann Neurol 2018;83:166–77.OpenUrlCrossRefPubMed ↵Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 2016;15:391–404.OpenUrlCrossRefPubMed ↵Li L, Sun L, Du R, et al. Application of the 2016 diagnostic approach for autoimmune encephalitis from Lancet Neurology to Chinese patients. BMC Neurol 2017;17:195.OpenUrl ↵Höftberger R, Titulaer MJ, Sabater L, et al. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology 2013;81:1500–6.OpenUrlCrossRefPubMed ↵Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776–85.OpenUrlCrossRefPubMed ↵Honnorat J, Didelot A, Karantoni E, et al. Autoimmune limbic encephalopathy and anti-Hu antibodies in children without cancer. Neurology 2013;80:2226–32.OpenUrlCrossRefPubMed ↵Graus F, Escudero D, Oleaga L, et al. Syndrome and outcome of antibody-negative limbic encephalitis [published erratum in Eur J Neurol 2018;25:1303]. Eur J Neurol 2018;25:1011–6.OpenUrl ↵van Sonderen A, Ariño H, Petit-Pedrol M, et al. The clinical spectrum of Caspr2 antibody-associated disease. Neurology 2016;87:521–8.OpenUrl ↵Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011;69:892–900.OpenUrlCrossRefPubMed ↵van Sonderen A, Thijs RD, Coenders EC, et al. Anti-LGI1 encephalitis: clinical syndrome and long-term follow-up. Neurology 2016;87:1449–56.OpenUrl Aurangzeb S, Symmonds M, Knight RK, et al. LGI1-antibody encephalitis is characterised by frequent, multifocal clinical and subclinical seizures. Seizure 2017;50:14–7.OpenUrl ↵Gadoth A, Pittock SJ, Dubey D, et al. Expanded phenotypes and outcomes among 256 LGI1/CASPR2-IgG-positive patients. Ann Neurol 2017;82:79–92.OpenUrl ↵Baumgartner A, Rauer S, Hottenrott T, et al. Admission diagnoses of patients later diagnosed with autoimmune encephalitis. J Neurol 2019;266:124–32.OpenUrl ↵Granerod J, Ambrose HE, Davies NW, et al.UK Health Protection Agency (HPA) Aetiology of Encephalitis Study Group. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis 2010;10:835–44.OpenUrlCrossRefPubMed ↵Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010;9:67–76.OpenUrlCrossRefPubMed Höftberger R, van Sonderen A, Leypoldt F, et al. Encephalitis and AMPA receptor antibodies: novel findings in a case series of 22 patients. Neurology 2015;84: 2403–12.OpenUrlPubMed Laurido-Soto O, Brier MR, Simon LE, et al. Patient characteristics and outcome associations in AMPA receptor encephalitis. J Neurol 2019;266:450–60.OpenUrl ↵Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157–65.OpenUrlCrossRefPubMed Spatola M, Sabater L, Planagumà J, et al. Encephalitis with mGluR5 antibodies: symptoms and antibody effects. Neurology 2018;90:e1964–72.OpenUrl Gresa-Arribas N, Planagumà J, Petit-Pedrol M, et al. Human neurexin-3α antibodies associate with encephalitis and alter synapse development. Neurology 2016;86:2235–42.OpenUrl Graus F, Keime-Guibert F, Reñe R, et al. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain 2001;124:1138–48.OpenUrlCrossRefPubMed Dalmau J, Graus F, Villarejo A, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain 2004;127:1831–44.OpenUrlCrossRefPubMed Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol 2015;72: 874–81.OpenUrl Pittock SJ, Lucchinetti CF, Parisi JE, et al. Amphiphysin autoimmunity: paraneoplastic accompaniments. Ann Neurol 2005;58:96–107.OpenUrlCrossRefPubMed Yu Z, Kryzer TJ, Griesmann GE, et al. CRMP-5 neuronal autoantibody: marker of lung cancer and thymoma-related autoimmunity. Ann Neurol 2001; 49: 146–54.OpenUrlCrossRefPubMed Do L-D, Chanson E, Desestret V, et al. Characteristics in limbic encephalitis with anti–adenylate kinase 5 autoantibodies. Neurology 2017;88:514–24.OpenUrl ↵Newey CR, Sarwal A, Hantus S. [(18)F]-fluoro-deoxy-glucose positron emission tomography scan should be obtained early in cases of autoimmune encephalitis. Autoimmune Dis 2016;2016:9450452. ↵Chow FC, Glaser CA, Sheriff H, et al. Use of clinical and neuroimaging characteristics to distinguish temporal lobe herpes simplex encephalitis from its mimics. Clin Infect Dis 2015;60:1377–83.OpenUrlCrossRefPubMed ↵Oyanguren B, Sánchez V, González FJ, et al. Limbic encephalitis: a clinical-radiological comparison between herpetic and autoimmune etiologies. Eur J Neurol 2013;20:1566–70.OpenUrlCrossRefPubMed ↵Budhram A, Silverman M, Burneo JG. Neurosyphilis mimicking autoimmune encephalitis in a 52-year-old man. CMAJ 2017;189:E962–5.OpenUrlFREE Full Text ↵Vogrig A, Joubert B, Ducray F, et al. Glioblastoma as differential diagnosis of autoimmune encephalitis. J Neurol 2018;265:669–77.OpenUrl ↵Kim JA, Chung JI, Yoon PH, et al. Transient MR signal changes in patients with generalized tonicoclonic seizure or status epilepticus: periictal diffusion-weighted imaging. AJNR Am J Neuroradiol 2001;22:1149–60. ↵Hosoki S, Satoi H, Matsumoto S. Bilateral hippocampal infarction mimicking limbic encephalitis. Intern Med 2018;57:911–2.OpenUrl ↵Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004;127:701–12.OpenUrlCrossRefPubMed ↵Steriade C, Moosa ANV, Hantus S, et al. Electroclinical features of seizures associated with autoimmune encephalitis. Seizure 2018;60:198–204.OpenUrl ↵Pessa ME, Janes F, Gigli GL. Electroencephalographic evaluation for early diagnosis of limbic encephalitis. Clin EEG Neurosci 2016;47:207–10.OpenUrlCrossRefPubMed ↵Malter MP, Elger CE, Surges R. Diagnostic value of CSF findings in antibody-associated limbic and anti-NMDAR-encephalitis. Seizure 2013;22:136–40.OpenUrlCrossRefPubMed ↵Weil AA, Glaser CA, Amad Z, et al. Patients with suspected herpes simplex encephalitis: rethinking an initial negative polymerase chain reaction result. Clin Infect Dis 2002;34:1154–7.OpenUrlCrossRefPubMed ↵Graus F, Delattre JY, Antoine JC, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry 2004;75:1135–40. ↵Lancaster E. Paraneoplastic disorders. Continuum (Minneap Minn) 2015; 21 (Neuro-oncology):452–75.OpenUrl ↵Voltz R, Gultekin SH, Rosenfeld MR, et al. A serologic marker of paraneoplastic limbic and brain-stem encephalitis in patients with testicular cancer. N Engl J Med 1999;340:1788–95.OpenUrlCrossRefPubMed ↵Saiz A, Blanco Y, Sabater L, et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain 2008;131:2553–63.OpenUrlCrossRefPubMed ↵Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008;7:1091–8.OpenUrlCrossRefPubMed ↵van Sonderen A, Petit-Pedrol M, Dalmau J, et al. The value of LGI1, Caspr2 and voltage-gated potassium channel antibodies in encephalitis. Nat Rev Neurol 2017;13:290–301.OpenUrl ↵Pellkofer HL, Kuempfel T, Jacobson L, et al. Non-paraneoplastic limbic encephalitis associated with NMDAR and VGKC antibodies. J Neurol Neurosurg Psychiatry 2010;81:1407–8.OpenUrlFREE Full Text ↵Titulaer MJ, Soffietti R, Dalmau J, et al.European Federation of Neurological Societies. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol 2011;18:19–e3.OpenUrlCrossRefPubMed ↵Dalmau J, Furneaux HM, Gralla RJ, et al. Detection of the anti-Hu antibody in the serum of patients with small cell lung cancer — a quantitative western blot analysis. Ann Neurol 1990;27:544–52.OpenUrlCrossRefPubMed ↵Budhram A, Nicolle MW, Yang L. The positive predictive value of onconeural antibody testing: a retrospective review. Can J Neurol Sci 2018;45:577–9.OpenUrl
|
Objective To report the clinical features and long-term outcome of 22 newly diagnosed paraneoplastic patients with GABAB receptor antibodies (GABABR-Abs).
|
|